
はじめに
脂質管理の歴史において、Proprotein Convertase Subtilisin/Kexin type 9;PCSK9の発見と阻害薬の開発は、最大級のイノベーションです。しかし、強力なLDLコレステロール(LDL-C)低下作用を持つ既存のモノクローナル抗体やsiRNA製剤は、すべて注射薬という形態をとらざるを得ませんでした。なぜなら、PCSK9とLDL受容体(LDLR)の結合面は非常に平坦かつ広大であり、従来の低分子化合物で結合を直接ブロックすることは創薬不可能(undruggable)と考えられてきたからです。
この膠着状態を打ち破ったのが、新規の経口低分子PCSK9阻害薬ラロプロブスタット(Laroprovstat; AZD0780)です。本論文は、従来の「結合阻害」ではなく「ドメイン安定化によるトラフィッキング制御」というまったく新しい分子生物学的アプローチにより、口頭投与可能な低分子でありながら、注射薬に匹敵する驚異的なLDL-C低下作用を示した最新のフェーズ1臨床試験のパラダイムシフトの実証記録です。
第1相試験(Phase 1 Trial)は、「この新薬候補は、人間の体にどれくらい安全に受け入れられ、どのように体内を巡るのか」を、少人数の被験者で科学的かつ厳格にサンプリング・検証する、創薬プロセスの極めて重要な足がかりとなる試験です 。
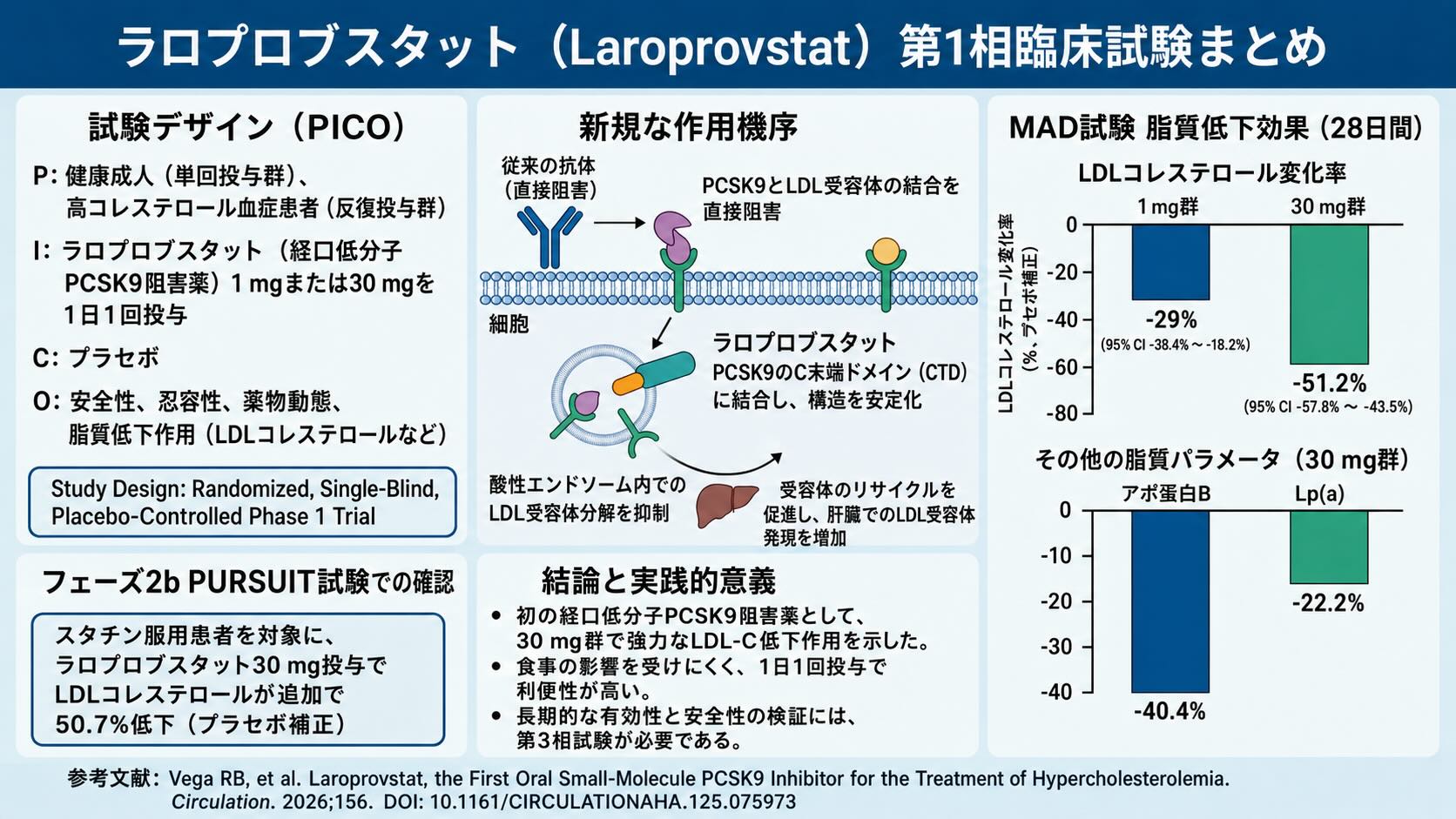
臨床研究プロトコール概要(PICO)と研究デザイン
本論文で報告された臨床試験の全体像を、医科学的視点から簡潔に整理します。
- P(Patient / 対象):
- 単回投与(SAD)試験:LDL-Cが70 mg/dL以上190 mg/dL以下の健康成人、計40名
- 複数回投与(MAD)試験:脂質低下療法の治療歴がない(treatment-naive)、スクリーニング時の空腹時LDL-Cが100 mg/dL以上190 mg/dL以下の高コレステロール血症患者、計62名
- I(Intervention / 介入):
- 単回投与試験:ラロプロブスタット30 mg、60 mg、120 mg、200 mg、400 mgの空腹時単回経口投与(120 mgコホートでは高脂肪食後の食後投与も実施)
- 複数回投与試験:ロスバスタチン20 mgを3週間先行投与(ランイン期間)し、LDL-Cが30%以上減少した患者に対して、ラロプロブスタット1 mgまたは30 mgを1日1回28日間経口投与
- C(Comparison / 比較): プラセボ投与群
- O(Outcome / 成果): 主要評価項目は安全性および忍容性。副次評価項目は、単回・定常状態におけるラロプロブスタットの血漿中薬物動態(PK)パラメータ、ならびにPCSK9およびLDL-Cレベルにおよぼす薬力学(PD)的影響。
- 研究デザイン: ランダム化、単盲検、プラセボ対照、第1相試験(SAD試験およびMAD試験)
分子生物学的視点
触媒ドメイン vs C末端ドメイン
ラロプロブスタットの真の真価は、既存の抗体医薬との決定的な「作用機序の新規性」にあります。既存の注射用PCSK9モノクローナル抗体は、PCSK9の触媒ドメインに結合し、LDLRとの物理的な結合そのものを阻害します。
これに対してラロプロブスタットは、結晶構造解析(PDBアクセッションコード: 9HZ3)により、PCSK9のC末端ドメイン(CTD)に存在する、独自の小さな結合ポケット(M2モジュールとM3モジュールの間)に特異的に結合することが判明しました。
細胞表面でのPCSK9とLDLRの結合を邪魔しない
分子生物学的な詳細を追うと、ラロプロブスタットとPCSK9のCTDとの間には、Val589およびSer636との水素結合トライアド(3つ組)をはじめ、Ala637やGly640との水素結合、Ser564およびHis591を介した水分子媒介型水素結合、さらにはGln587との非古典的水素結合など、緻密な相互作用ネットワークが形成されています。表面プラズモン共鳴(SPR)試験において、ラロプロブスタットはフルレングスのヒトPCSK9に対して2.30 +- 0.25 nmol/Lという極めて高い親和性(KD値)で結合します。しかし驚くべきことに、この化合物が結合した状態でも、PCSK9とLDLRの結合親和性は、中性環境(pH 7.4)および酸性環境(pH 5.6)のいずれにおいても全く変化しません。つまり、細胞表面でのPCSK9とLDLRの結合を「邪魔しない」のです。
細胞内の「エンドソーム」における物質輸送制御
では、なぜLDL-Cが下がるのでしょうか。その秘密は細胞内の「エンドソーム」におけるトラフィッキング(物質輸送)の制御にあります。
通常、細胞表面でLDLRに結合したPCSK9は、複合体を形成したままエンドソームへと内包(エンドサイトーシス)されます。エンドソーム内部はpH 5.6〜6.5程度の酸性環境に保たれており、PCSK9のCTDはこの酸性を感知すると構造的な柔軟性を変化させ、複合体をリソソームでの分解ルートへと強制的に誘導します。これがLDLRを破滅に導く酸性の罠です。
ラロプロブスタットがCTDのポケットに結合すると、示差走査熱量測定(DSC)で示されたように、CTDの熱力学的安定性と柔軟性が劇的に変化します。酸性環境下(pH 5.6)において、未結合のCTDに比べて melting temperature(Tm:変性温度)が顕著に上昇し、カチカチに構造が固定されるのです。水素デウテリウム交換質量分析(HDX-MS)でも、酸性条件下(pH 6.5)でラロプロブスタットが結合している部位の構造が高度にリジッド(硬固)になっていることが視覚的に証明されました。
このように構造がカチカチに安定化されたPCSK9は、酸性エンドソーム内に放り込まれても「リソソームへ運べ」という分解シグナルを出すことができなくなります。結果として、リソソームトラフィッキングおよび分解のプロセスが阻止され、LDLRは無傷のままエンドソームから離脱し、細胞膜表面へと再びリサイクル(再循環)されていきます。細胞表面のLDLRが劇的に増えれば、血液中からのLDL粒子のクリアランスが爆発的に向上します。これが、結合を阻害せずに構造を安定化させて標的を救う、ラロプロブスタット独自の洗練されたサイエンスです。
臨床データが語る圧倒的なインパクト:スタチン上乗せで80%の衝撃
この緻密な分子生物学的メカニズムが、実際のヒトの体内(in vivo)でどれほど強力な臨床効果をもたらしたのか、詳細な数値を検証します。
複数回投与(MAD)試験
まず複数回投与(MAD)試験に先立ち、対象となった高コレステロール血症患者は、ロスバスタチン20 mgによる3週間のランイン治療(Run-in period;準備・前治療の期間)を受けました。スタチン単独の段階で、患者のLDL-Cレベルはすでに untreated baseline(未治療ベースライン)から平均55%減少していました。この「スタチンで十分に叩いた状態」から、ラロプロブスタット30 mgを1日1回、28日間上乗せ投与する併用療法が行われました。
その結果、投与開始から迅速に脂質レベルは低下し、投与14日目までに定常状態(定常的な減少)に達しました。28日目におけるプラセボ補正後(対プラセボ)のLDL-C変化率は、ラロプロブスタット1 mg群で-29%(95%信頼区間: -38.4% 〜 -18.2%)、そしてラロプロブスタット30 mg群では実に-51.2%(95%信頼区間: -57.8% 〜 -43.5%)という劇的な追加減少を記録しました。この結果、ロスバスタチン20 mgとラロプロブスタット30 mgの強力なコンビネーションにより、未治療ベースラインからの総LDL-C減少率は、およそ80%という驚異的な領域にまで達したのです。既存の注射用PCSK9阻害薬を上乗せした場合と同等以上の脂質低下効果が、小さな「錠剤」を毎日飲むだけで達成されました。
ApoB, Lp(a), トリグリセリド
さらに、この臨床的インパクトはLDL-Cだけにとどまりません。プラセボ補正後のデータにおいて、動脈硬化を強力に促進するリスク因子であるアポリポタンパク質B(ApoB)は、1 mg群で-24.7%、30 mg群で-40.4%と有意に減少しました。
また、既存の治療薬ではコントロールが極めて難しいとされる独立した心血管リスク因子、リポタンパク質(a)[Lp(a)]についても、1 mg群で-12.1%、30 mg群で-22.2%(95%信頼区間: -35.2% 〜 -6.5%)という明確な減少効果が示されました。その他、総コレステロールは-25.4%、nonHDL-Cは-44.6%、トリグリセリドは-19.6%と、包括的な脂質プロファイルの改善が確認されています(いずれも30 mg群のデータ)。
単回投与(SAD)試験;優れた薬物動態(PK)
薬物動態(PK)の観点からも、ラロプロブスタットは優れたプロファイルを示しています。単回投与(SAD)試験において、30 mgから400 mgの広い用量範囲でCmaxは完璧な用量比例性(パワーモデルにおける傾きパラメーター0.9837、90%信頼区間: 0.9168 〜 1.051)を示しました。血中半減期は約40時間と長く、この長い半減期が1日1回投与という簡便なレジメンを完全に支えています。
さらに重要なのは、食事による曝露量への影響です。120 mgを用いて高脂肪食後に投与したPK検証では、空腹時と比較した幾何平均比がAUCtで1.15(90%信頼区間: 1.11 〜 1.19)、Cmaxで1.06(90%信頼区間: 1.00 〜 1.13)であり、臨床的に意味のある曝露量の変化は見られませんでした。つまり、「食事の有無に関わらず、いつでも服用可能」という極めて高い患者利便性が実証されたと言えます。また、中国系背景を持つコホートとの比較においても、非アジア系被験者との間に臨床的に意味のある曝露量の差は認められませんでした。
臨床における安全性の検証と厳格な批判的吟味(Limitation)
日常の臨床現場、あるいは今後の展開を見据える上で、安全性シグナルの評価と試験デザインの限界点を批判的に吟味することは不可欠です。
有害事象の深刻度はすべて軽度から中等度
28日間のMAD試験において、ラロプロブスタットは全体として優れた忍容性を示しました。死亡例や重篤な有害事象(SAE)の報告はなく、有害事象の総発現率はラロプロブスタット群で34.2%(38例中13例)、プラセボ群で36.4%(22例中8例)であり、プラセボと比較して明確な増加傾向は認められませんでした。発現した有害事象の深刻度はすべて軽度から中等度でした。
治療中止例
しかし、詳細なデータ(Tables S4, S5)を吟味すると、有害事象による治療中止例が少数ながら発生している点に注意を向ける必要があります。ラロプロブスタット30 mg群の1名、およびプラセボ群の1名において、無症候性の非持続性心室頻拍(nonsustained ventricular tachycardia)による試験中止が記録されています。また、ラロプロブスタット1 mg群の1名において、肝機能指標であるアラニンアミノ転移酵素(ALT)が基準値上限の3倍を超えて上昇したため、投与が中止されました(なお、同症例においてビリルビンの上昇は伴っていません)。これらの中止例について、前者はプラセボ群でも発生している点、後者は低用量群でのみ発生している点から、現時点で一概に薬物との直接的な因果関係を断定することはできませんが、今後の拡大試験において細心の注意を払うべき安全性プールの指標となります。
初期の第1相臨床試験
そして、本研究における最大の限界点(Limitation)は、これが極めて初期の第1相臨床試験であるという事実そのものです。MAD試験におけるラロプロブスタット30 mg群の被験者数はわずか18例であり、統計的な母集団としては非常に小規模です。多様な合併症を持つ患者や、より高齢の患者層における安全性は未だ不透明です。また、連続投与期間が28日間という超短期であることも大きな制約です。動脈硬化性心疾患の予防管理は、実臨床においては数年、数十年という生涯にわたる長期の治療継続が前提となります。したがって、本剤が長期間にわたって安全に脂質低下作用を維持できるか、また、真の臨床アウトカムである「心血管イベント(心筋梗塞や脳卒中など)の発生抑制」に実際に寄与できるかという点については、本論文のデータだけでは結論づけることができません。
すでに論文内で言及されているフェーズ2b PURSUIT試験などの大規模データを経て、今後のフェーズ3試験による長期的な有効性と安全性のレジストリ確立が待たれる状況です。
第2相臨床試験も既に行われている
なお、この論文の中で言及されているフェーズ2b「PURSUIT試験」の結果の概要は、以下のとおりです 。
- 対象患者: すでに中強度または高強度のスタチン(コレステロールを下げる標準治療薬)を服用しているにもかかわらず、数値が十分に下がっていない脂質異常症の患者 。
- 投与量: ラロプロブスタット 30 mg を1日1回投与 。
- 結果(有効性): プラセボ(偽薬)群と比較して、LDLコレステロール(悪玉コレステロール)を追加で50.7%減少させた 。
この結果は、今回のフェーズ1試験(スタチン上乗せで51.2%減少)とほぼ完全に一致しています 。少人数での実験的なデータ(フェーズ1)が、より実臨床に近い大規模な患者集団(フェーズ2b)でも高い再現性をもって実証されたことを意味しています 。
明日からの臨床と研究に活かす実証的アプローチ
本論文の知見は、単なる未来の新薬候補の紹介にとどまらず、最前線に立つ医療人や研究者の明日の行動に変革を促す重要な示唆を含んでいます。読者が明日から実践できる、あるいは思考に活かせるポイントを3つの軸で提示します。
超強力な脂質管理(LDL-C 30 mg/dL未満)の未来を見据えた患者選択のシミュレーション
現在の動脈硬化性心血管疾患(ASCVD)ガイドラインでは、極めて高いリスクを持つ患者に対して、かつてないほど厳格なLDL-C目標値(例えば55 mg/dL未満、あるいは二次予防においてさらなる低値)が推奨されています。実臨床において、高用量スタチンとエゼチミブを併用しても目標値に届かない「スタチン抵抗性」の患者に直面したとき、将来的に経口のPCSK9阻害薬が登場した場合、どのようなタイムラインで追加すべきか、今から具体的な症例をベースに治療シミュレーションを頭の中で組み立ててください。注射薬を自己注射することへの心理的・経済的ハードルが高く、PCSK9阻害薬の恩恵を受けられずにいる潜在的な高リスク患者リストを、明日のカルテからピックアップし始める動機になります。
「食事制限なし・1日1回」がもたらすアドヒアランス低下リスクの先回り評価
ラロプロブスタットの大きな強みは、高脂肪食の後であっても空腹時であっても曝露量が変化しないという、食事制限のなさです。これは患者のアドヒアランス(服薬順守)を大幅に向上させますが、一方で「いつでも飲んで良い」という自由度は、服薬タイミングのルーティン化を妨げ、飲み忘れを誘発するリスクも孕んでいます。明日の外来から、既存の脂質低下薬(スタチンなど)を内服している患者に対し、食事のタイミングや1日の生活スケジュールの中で「どの時間帯が最も確実に、飲み忘れなく内服できているか」の行動パターンを詳細にヒアリングしてください。将来、強力な経口新薬が上市された際に、最高のパフォーマンスを発揮させるための服薬指導ノウハウが今から蓄積されます。
分子生物学的「ドメイン安定化」という次世代創薬視点の獲得
本論文に示された「標的タンパク質の結合面をブロックするのではなく、別のドメインを安定化させて細胞内リサイクルルートを変える」という手法は、循環器領域のみならず、あらゆる創薬科学における最先端のトレンドです。明日からの学術的リサーチや論文抄読会において、他の「創薬不可能」とされてきた標的タンパク質(例えば特定の腫瘍マーカーや受容体)に対しても、同様の構造安定化やトラフィッキング制御を試みている基礎研究がないか、アンテナの感度を大きく広げてください。本論文を通じて得た「ドメイン安定化によるリサイクル制御」という概念的なレンズは、最新のバイオロジーを読み解く強力な武器となります。
経口低分子PCSK9阻害薬ラロプロブスタットは、強力なスタチン治療に1錠を加えるだけで、合計80%のLDL-C低下を食事制限なく達成するという、これまでの脂質管理の常識を覆す未来の姿を鮮明に描き出しました。このサイエンスの進歩がもたらすパラダイムシフトの波を正確に捉え、明日の医療、そして研究へと還元していきましょう。
参考文献
Vega RB, O’Mahony G, Barbour AM, Yu H, Knochel J, Brengdahl J, Hochdorfer T, Bergenholm L, Toppner Carlsson E, Ahnmark A, Rye Underwood C, Rudvik A, Carter D, Laru J, Gutgsell A, Twaddle L, Garkaviy P, Bogstedt A, Hurt-Camejo E, Miliotis T, Ryaboshapkina M, Hober A, Hubbard B, Serrano-Wu M, Kaushik V, Geschwindner S, McCarthy MC, Linden D, Rosenmeier JB. Laroprovstat, the First Oral Small-Molecule PCSK9 Inhibitor for the Treatment of Hypercholesterolemia: Results From a Randomized, Single-Blind, Placebo-Controlled Phase 1 Trial in Treatment-Naive Patients. Circulation. 2026;156. DOI: 10.1161/CIRCULATIONAHA.125.075973.
おまけ(難しい):LDLコレステロール、PCSK9、既存のPCSK9阻害薬、ラロプロブスタットがLDL受容体に結合した際の挙動
LDL受容体(LDLR)に各物質が結合した際の挙動について、細胞内での運命、最終的なLDLコレステロール(LDL-C)への影響、および薬理学的なメカニズムの違いを表で整理しました。
5つのパターンの比較一覧表
| パターン | 細胞表面での結合状態 | エンドソーム内(酸性)での挙動 | LDL受容体の運命 | 血液中のLDL-C濃度 | 薬理学的なメカニズムの分類 |
| ① LDL単独 | LDL粒子が受容体の先端(リガンド結合ドメイン)に結合する。 | 酸性環境によって受容体の形が変わり、LDL粒子をパッと離す。 | 細胞表面へリサイクル(生存) 次の回収へ向かう(約100回反復)。 | スムーズに回収されて下がる。 | 生理的な正常ルート(基準となるリサイクル機構) |
| ② PCSK9単独 | 受容体の根元(EGF-Aドメイン)にPCSK9が単独で結合する(先端は空席)。 | 酸性になってもPCSK9は離れない。CTDが変形し破壊シグナルを出す 。 | リソソームへ送られ一緒に破壊(消滅) | 次のLDLを回収する受容体が減るため上がる 。 | 標的タンパク質分解ルート(生理的な受容体数の調節) |
| ③ LDL + PCSK9 | 受容体の先端にLDL、根元にPCSK9が同時に結合する。 | 酸性になってもPCSK9は離れない。CTDが変形し破壊シグナルを出す 。 | リソソームへ送られ一緒に破壊(消滅) | 同乗していたLDLごと破壊され、受容体も減るため上がる 。 | 標的タンパク質分解ルート(コレステロール回収効率の低下) |
| ④ LDL + PCSK9 + 既存のPCSK9i(レパーサ等) | 抗体がPCSK9の結合面を覆うため、PCSK9は受容体に結合できない 。LDLのみが結合する。 | 結合できなかったPCSK9は血中に残り、受容体側は①(LDL単独)と同じ挙動をとる。 | 細胞表面へリサイクル(生存) | 受容体が保護されて豊富にあるため劇的に下がる 。 | 中和抗体 / PPI(タンパク質間相互作用)阻害薬 細胞外での物理的結合ブロック |
| ⑤ LDL + PCSK9 + ラロプロブスタット | 薬はPCSK9のC末端(CTD)に結合するが、受容体との結合は邪魔しないためすべて同時に結合する 。 | 薬がCTDをカチカチに固めるため、酸性になっても破壊シグナルを出せなくなる 。 | 細胞表面へリサイクル(生存) | 受容体がリサイクルされるため劇的に下がる 。 | 構造安定化 / トラフィッキング(輸送)制御薬 細胞内での機能特異的阻害 |
重要なポイントの補足
- 「荷物(LDL)」の有無に関わらない破壊: パターン②と③の比較から分かる通り、PCSK9が受容体を破壊に導くプロセスにおいて、LDL粒子が一緒に結合しているかどうかは関係ありません 。PCSK9が受容体にくっついた時点で、その受容体のリサイクルルートは閉ざされてしまいます 。
- ラロプロブスタット(⑤)の網羅性: ラロプロブスタットは、LDLが同乗しているパターン(③→⑤)だけでなく、PCSK9が単独で受容体を間引こうとして細胞内に入ってきたパターン(②)に対しても、酸性エンドソーム内で完全にその破壊シグナルを無効化し、受容体を安全にリサイクルルートへ逃がすことができます 。
- 「結合」の有無: 既存の注射薬(③)は、細胞表面でPCSK9が受容体にくっつくこと自体を絶対に許しません 。対してラロプロブスタット(④)は、結合すること自体は許容します 。
- 「解決」の場所: 既存の注射薬(③)は「細胞の外(血液中)」で問題を解決するのに対し 、ラロプロブスタット(④)は一旦すべてを細胞内に受け入れた上で、「細胞の中(エンドソーム)」で破壊シグナルを無効化して受容体を救出します 。

