
はじめに
動脈硬化性心血管疾患(atherosclerotic cardiovascular disease;ASCVD)の抑制において、LDLコレステロール(low-density lipoprotein–cholesterol ;LDL-C)の低減は「低ければ低いほど良い」というパラダイムが確立されています。しかし、強力なエビデンスを持つスタチンであっても、副作用による不耐容や効果不十分な症例が一定数存在します。この課題に対する分子生物学的な回答として登場したのが、世界初のATPクエン酸リアーゼ(ATP citrate lyase;ACL)阻害薬であるベンペド酸(Bempedoic Acid)です。本稿では、2024年のレビュー論文に基づき、その独自の薬理プロファイルと、臨床で避けては通れない薬物相互作用(drug–drug interactions ;DDI)の深淵を解き明かします。
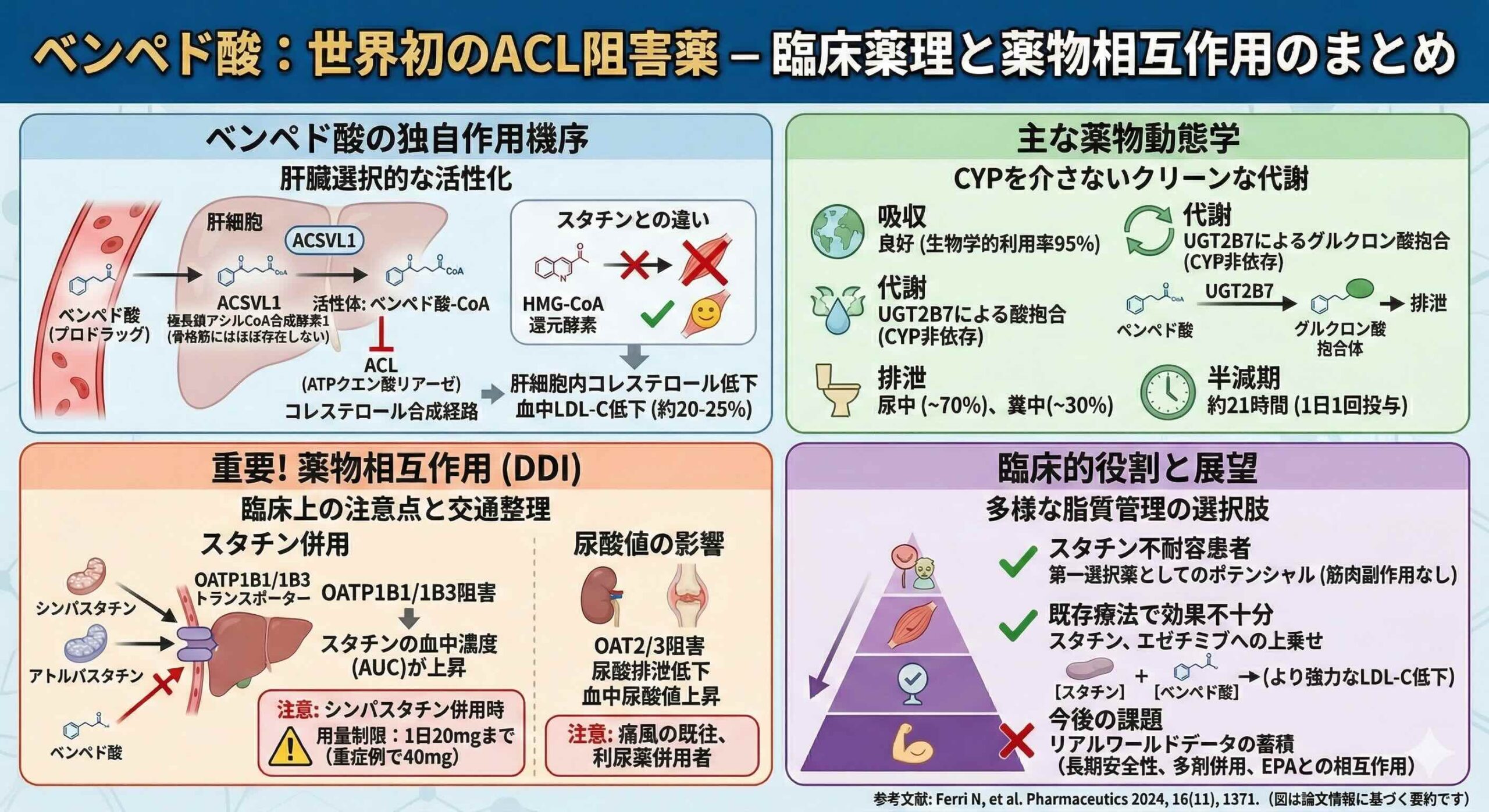
ベムペド酸は肝臓で選択的にコレステロール合成を阻害
ベムペド酸の最大の革新性は、その活性化プロセスに隠されています。スタチンが全身の細胞でHMG-CoA還元酵素を阻害するのに対し、ベムペド酸は特定の酵素が存在する組織でのみ牙を剥くプロドラッグです。
この薬剤を活性化するのは、極長鎖アシルCoA合成酵素1(very-long-chain acyl-CoA synthetase 1;ACSVL1)という酵素です。重要な事実は、このACSVL1が肝臓には豊富に発現している一方で、骨格筋にはほとんど存在しないという点です。この組織発現の乖離により、ベムペド酸は肝臓で選択的にコレステロール合成を阻害し、スタチンで頻発する筋肉関連副作用を回避するという、極めて合理的な設計がなされています。まさに、肝臓を狙い撃つ分子の芸術と言えるでしょう。
コレステロール合成阻害のメカニズム
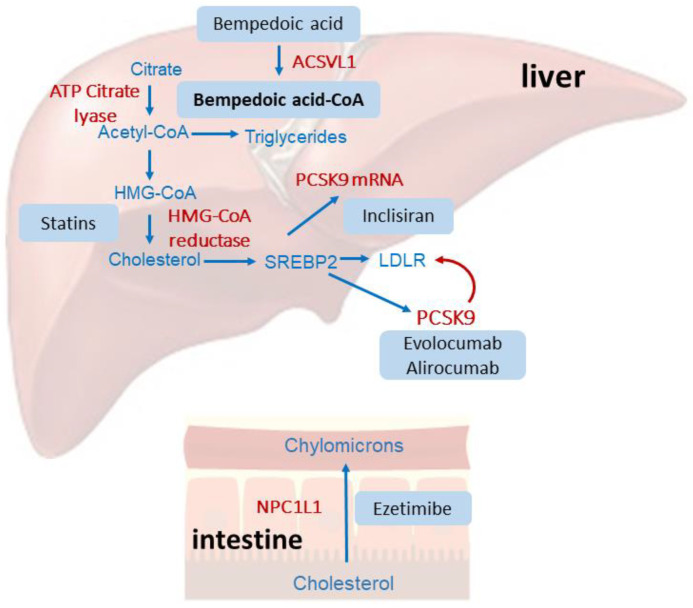
ベムペド酸が標的とするATPクエン酸リアーゼ(ATP citrate lyase;ACL)は、コレステロールおよび脂肪酸合成経路の初期段階を担う鍵酵素です。ベンペド酸は肝細胞内でACSVL1によって活性体であるベムペド酸-CoA(bempedoic acid–CoA)へと変換されます。この活性体は10µMというIC50値でACLを強力かつ選択的に阻害します。
この阻害により細胞内のアセチルCoA供給が断たれると、肝細胞はコレステロール不足を感知します。すると、転写因子であるステロール調節エレメント結合タンパク質2( sterol regulatory element-binding protein 2;SREBP2)が活性化され、LDL受容体の発現を増強します。結果として、血中のLDL-Cの取り込みが促進され、血清レベルが低下するのです。
臨床試験データでは、ベムペド酸単剤でLDL-Cを20%から25%、非HDLコレステロールを19%、アポリポタンパク質B(apoB)を15%、総コレステロールを16%減少させることが示されています。
薬物動態、CYPを介さない代謝経路
ベムペド酸の薬物動態は、多くの現代的な併用療法において非常に有利に働きます。経口投与後のバイオアベイラビリティは95%と極めて高く、食事の影響を受けません。最高血中濃度到達時間(Tmax)は3.5時間、半減期は19時間から21時間と、1日1回の投与に適した安定した挙動を示します。
特筆すべきは代謝経路です。ベムペド酸およびその代謝物は、薬物代謝の主要経路であるシトクロムP450(cytochrome P450 ; CYP450)の基質でも、誘導剤でも、阻害剤でもありません。主な代謝は、UGT2B7によるグルクロン酸抱合です。これにより、CYPを介した複雑な薬物相互作用の連鎖から解放されており、多くの併用薬を持つ高齢者や合併症患者において、処方の自由度を高めています。
薬物相互作用;スタチンとの併用の注意点
ベムペド酸自体はCYP代謝を受けませんが、トランスポーターに対する阻害能を持つため、Perpetrator(加害者)としての側面には細心の注意が必要です。
最も警戒すべきはスタチンとの相互作用です。ベムペド酸およびその抱合体は、肝取り込みトランスポーターであるOATP1B1およびOATP1B3を弱く阻害します。
ベムペド酸がOATP1B1/1B3を阻害するということは、同じ門を通ろうとするスタチンにとって「入り口が塞がれる」ことを意味します。肝臓はスタチンの主な作用部位ですが、同時に代謝の場でもあります。トランスポーターが阻害されると、スタチンは肝臓に入ることができず、結果として血液中に長時間留まることになります。
臨床試験において、ベムペド酸180mgの定常状態下でシンバスタチン40mgを単回投与したところ、シンバスタチン酸の露出量(AUC)が2倍に上昇することが確認されました。アトルバスタチン、プラバスタチン、ロスバスタチンにおいても1.4倍から1.5倍の上昇が認められています。
肝内での取り込みが制限されているためスタチン単体のコレステロール低下効果が強まることはありません。 血液中に滞留した高濃度のスタチンは本来の作用点ではない骨格筋などの他組織へと移行し、筋肉痛などの副作用リスクを有意に高める結果を招きます。
(スタチンはコレステロール合成経路の「下流(HMG-CoA還元酵素)」を阻害し、ベムペド酸は「上流(ACL)」を阻害します。異なる段階を同時にブロックするため、たとえスタチン単体の効率が多少落ちたとしても、ベンペド酸による上流の阻害効果がそれを遥かに上回り、肝細胞内コレステロールを減らす力は、スタチン単剤よりも飛躍的に高まります。)
このため、ベムペド酸とシンバスタチンを併用する場合、シンバスタチンの用量は1日20mg(重症例で40mg)に制限する必要があります。一方で、アトルバスタチンやロスバスタチンとの相互作用は臨床的に許容範囲内とされていますが、スタチン関連副作用の増強には常に監視が必要です。
腎トランスポーター阻害:尿酸値上昇
ベムペド酸の副作用として知られる尿酸値の上昇も、トランスポーター相互作用によって説明されます。ベムペド酸は腎臓の尿酸トランスポーターであるOAT2およびOAT3を阻害します。これにより、尿酸の排泄が抑制され、血中尿酸値の可逆的な上昇を招きます。
特に、ループ利尿薬やチアジド系利尿薬を使用している患者では、尿酸値のさらなる上昇と痛風発症のリスクが高まります。また、腎機能を評価する際の指標であるクレアチニン値も、同様の機序でわずかに上昇する可能性があります。これらの変化は軽微かつ可逆的ですが、慢性腎臓病(CKD)患者や痛風の既往がある患者では、定期的な生化学検査が不可欠です。
未知の領域とVictim(被害者)としての側面
ベムペド酸がVictim(被害者)となるケースは限定的ですが、UGTおよびOAT3の強力な阻害薬であるプロベネシド(商品名:ベネシッド)との併用には注意が必要です。プロベネシドの併用により、ベンペド酸の露出量は1.7倍、活性代謝物の露出量は1.9倍に増加します。これ自体は直ちに毒性につながるレベルではありませんが、薬理効果が増強される可能性があります。
また、興味深い理論的相互作用として、高純度EPA製剤(イコサペント酸エチル)(商品名:エパデールなど)との競合が挙げられます。EPAもベンペド酸と同様に肝臓のACSVL1の基質となるため、両者を併用した場合、ベンペド酸の活性化が理論上阻害される可能性が指摘されています。これが臨床的な効果減弱につながるかどうかは、今後のさらなる研究が待たれる領域です。
本研究の新規性と臨床的限界(Limitation)
本研究の新規性は、ベンペド酸の分子生物学的な組織選択性と、最新の臨床試験およびリアルワールドデータに基づく包括的なDDIマップを提示した点にあります。特に、スタチン不耐容患者における心血管イベント抑制効果を証明したCLEAR Outcomes試験の結果を背景に、実臨床での処方優先順位を整理した意義は大きいです。
しかし、本研究にはいくつかの限界も存在します。
第一に、ベムペド酸と最新の併用療法、例えばPCSK9阻害薬やインクリシランとの直接的なDDIデータは依然として限られています。
第二に、高度な腎機能障害患者や肝機能障害患者における長期的な安全性データがまだ十分とは言えません。
第三に、コレステロール以外の脂質パラメータや、イコサペント酸エチルとの相互作用については、あくまで分子理論に基づく推測の域を出ていない部分があります。
明日から実践できる行動:ベンペド酸を使いこなすための指針
本論文の知見を明日の臨床に活かすため、以下の3点を推奨します。
第一に、スタチン不耐容、あるいはスタチン最大用量でもLDL-C目標値に達しない患者において、ベムペド酸を早期に検討してください。その際、筋肉痛のリスクが極めて低いという分子生物学的根拠を説明することで、患者の服薬アドヒアランス向上につなげることが可能です。
第二に、シンバスタチンを服用中の患者にベムペド酸を追加する場合は、必ずシンバスタチンの用量を確認し、必要に応じて20mg以下への減量、あるいはアトルバスタチンやロスバスタチンへの切り替えを検討してください。
第三に、ベムペド酸開始前および開始後には、ベースラインの尿酸値を確認してください。特に利尿薬を併用している患者や、高齢者、CKD合併例では、投与開始から数週間後の尿酸値モニタリングをルーチン化することで、痛風発作を未然に防ぐことができます。
ベムペド酸は、単なる「スタチンの代替品」ではありません。肝臓選択的な代謝活性化の機序を持つ、次世代の脂質低下療法の新たな選択肢となる薬剤です。その薬理学的特性を深く理解し、DDIを適切に管理することで、私たちはより安全で確実な心血管イベント抑制を達成できるはずです。
参考文献
Ferri, N.; Colombo, E.; Corsini, A. Bempedoic Acid, the First-in-Class Oral ATP Citrate Lyase Inhibitor with Hypocholesterolemic Activity: Clinical Pharmacology and Drug-Drug Interactions. Pharmaceutics 2024, 16, 1371. https://doi.org/10.3390/pharmaceutics16111371

