
はじめに
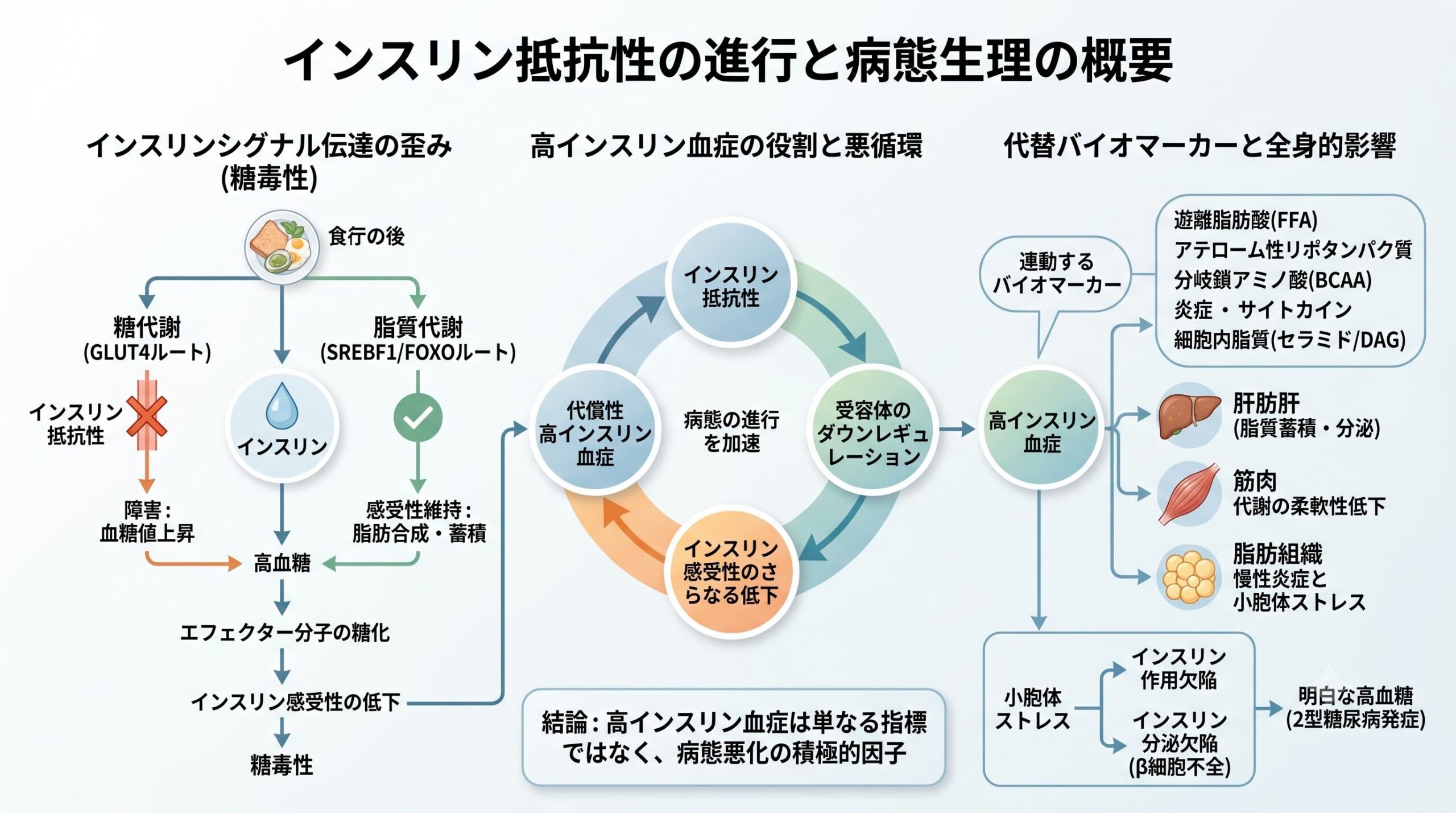
2型糖尿病の根底に存在するインスリン抵抗性は、従来、インスリン受容体から下流へのシグナルが一様に遮断された状態として理解されてきました。しかし、この単純なモデルでは、患者の体内において血糖値が上昇しているにもかかわらず、肝臓での脂質合成やアテローム性リポタンパク質の分泌がむしろ亢進しているという臨床的パラドックスを説明できません。
本レビュー論文で示された最新の知見は、この謎に対して明快なパラダイムシフトを提示しています。インスリン抵抗性の本質は、シグナル全体の単純なブロックではなく、特定の伝達経路のみが障害され、他の経路は温存される「選択的シグナル障害」にあります。そして、この不均衡をさらに悪化させる主犯こそが、低下した感受性を補おうとして生体が分泌する過剰なインスリン、すなわち高インスリン血症そのものであるという衝撃的な事実です。本稿では、最新の分子生物学的知見を織り交ぜながら、インスリン抵抗性の真の姿に迫ります。
精緻を極めるインスリンシグナル伝達と停止の分子生物学
インスリンシグナル伝達
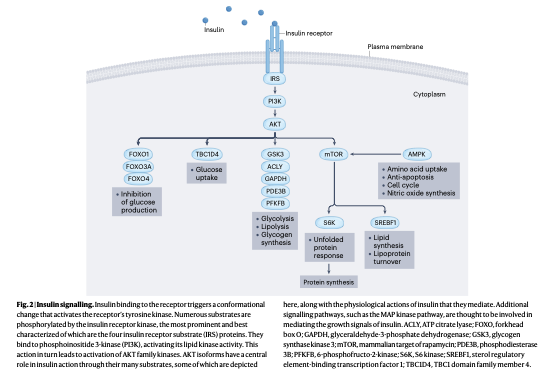
インスリン受容体は、刺激を受ける前からすでにプレアセンブルされたホモ二量体として細胞膜上に存在するという、チロシンキナーゼ型受容体の中でもユニークな特徴を持っています。クライオ電子顕微鏡による構造解析により、この受容体は最大4分子のインスリンと結合可能ですが、わずか1分子のインスリンが結合するだけで完全な活性化トリガーが引かれることが判明しています。
インスリンの結合に伴い受容体は立体構造を変化させ、ベータサブユニットに位置するリシン1030残基がATPと結合してオールスター的(アロステリック)にチロシンキナーゼを活性化します。このチロシンリン酸化シグナルは、4つのアダプタータンパク質であるIRS1からIRS4(insulin receptor substrates)を介して脂質キナーゼであるPI3Kに伝わり、最終的にセリン・スレオニンキナーゼであるAKTを活性化します。
活性化したAKTは、極めて多岐にわたる生物学的プロセスを制御します。
第1に、転写因子であるFOXO1、FOXO3a、FOXO4をリン酸化して核外へ排除し、標的遺伝子の転写を抑制することで、肝臓における糖新生を強力に抑えます。
第2に、筋肉細胞や脂肪細胞においてTBC1D4(AS160とも呼ばれます)をリン酸化し、糖輸送体であるGLUT4を含有する小胞の細胞膜への移行を促して糖の取り込みを促進します。
第3に、GSK3、ACLY、GAPDH、PDE3B、PFKFBなどの代謝酵素群を制御し、糖新生の抑制、糖分解およびグリコーゲン合成の促進、脂肪分解の抑制を素早く実行します。
第4に、mTOR、S6K、AMPKを介してタンパク質翻訳を調節するとともに、転写因子SREBF1を介した脂質合成(デノボ脂質合成)を亢進させます。
インスリンシグナル停止
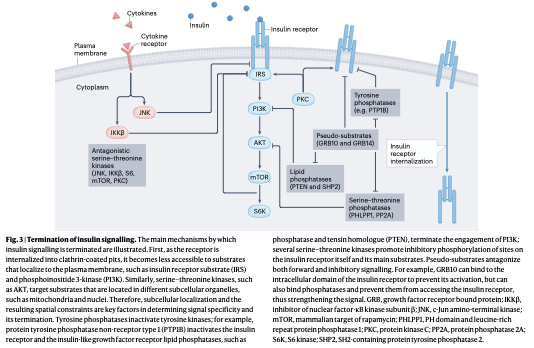
一方で、過剰なインスリン作用による致命的な低血糖を防ぐため、シグナルの遮断機構もまた極めて緻密かつ多重に構築されています。活性化した受容体は、クラスリン被覆ピットを介して速やかに細胞内へと内飲(エンドサイトーシス)されます。また、チロシンホスファターゼ(PTPN1など)や脂質ホスファターゼ(PTENやSHP2)、セリン・スレオニンホスファターゼ(PHLPP1やPP2A)が各シグナル分子を脱リン酸化して不活性化します。
さらに、GRB10やGRB14といった疑似基質タンパク質は、受容体のキナーゼドメインに直接結合して活性化を防ぐだけでなく、ホスファターゼの接近をブロックしてシグナルを安定化させるという、両刃の剣としての複雑な役割を担っています。IRS1のセリン307残基におけるリン酸化は、培養細胞レベルではシグナルを抑制しますが、生体内(in vivo)では逆にインスリン感受性を高めて糖尿病発症を防ぐという極めて難解なパラドックスが確認されており、シグナルの終息機構が単純なオフスイッチではないことを物語っています。
既存説への挑戦:DAG・PKC仮説の揺らぎと原因と結果の再考
これまで、脂質がインスリン抵抗性を引き起こすメカニズムとして、細胞内に蓄積したジアシルグリセロール(diacylglycerol;DAG)が非定型プロテインキナーゼC(Protein Kinase C;PKC)を活性化し、これがインスリン受容体のスレオニン1160残基をリン酸化してキナーゼ活性を抑制するという「DAG・PKC仮説」が広く支持されてきました。
しかし、本レビューはヒトの遺伝学研究およびトランスレーショナルデータに基づき、この仮説に対して大きな疑問を投げかけています。その最も明確な反証が、PNPLA3遺伝子やATGL遺伝子の変異を持つ患者群のデータです。これらの患者では、肝臓内に極めて大量のDAGやトリグリセリドが蓄積しているにもかかわらず、インスリン抵抗性を合併していません。
また、遺伝子改変マウスを用いた研究でも、AKT2のノックアウトは肝臓の脂質蓄積を防ぐ一方で、FOXO遺伝子群をトリプルノックアウトして恒常的にインスリン感受性を高めたマウスでは、逆に脂質蓄積が誘発されることが示されています。
これらの強固な事実から論理的に導き出される結論は、「脂質の蓄積がインスリン抵抗性を引き起こす」という従来の因果関係の否定です。むしろ、「インスリン抵抗性によって生じるシグナル伝達の不均衡(糖代謝シグナルの障害と脂質合成シグナルの温存)の結果として、二次的に細胞内へ脂質が蓄積する」という逆の因果関係が強く示唆されます。
高インスリン血症が引き起こす選択的インスリン抵抗性のメカニズム
なぜ、同一の細胞内でシグナルの障害と温存が同時に発生するのでしょうか。高インスリン血症がこの選択的インスリン感受性selective insulin sensitization 低下を引き起こす具体的なプロセスを、脂肪細胞と肝細胞の2つのモデルで検証します。
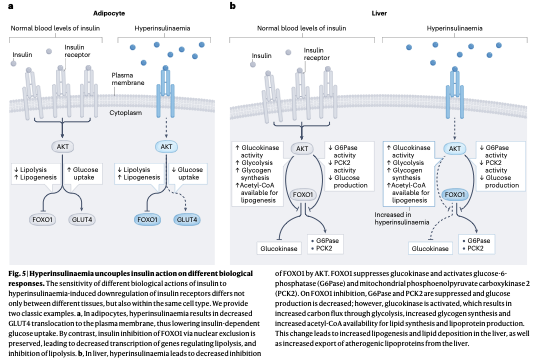
脂肪細胞における不均衡
正常なインスリン濃度下では、糖の取り込み(GLUT4移行)と脂肪分解の抑制(FOXO1核外排除)の双方が適切に制御されています。
しかし、高インスリン血症によって受容体数が減少すると、GLUT4の膜移行シグナルは極めて早期に枯渇し、細胞内への糖の取り込みは劇的に低下します。
これに対して、FOXO1を核から排除して脂肪分解を抑え、脂質合成を促進するシグナル伝達系は、受容体が減少した状態でも高い感受性を維持し続けます。
この結果、脂肪細胞は糖の取り込み障害を起こしつつも、脂肪の蓄積と合成だけをひたすら継続するという、選択的な抵抗性病態が完成します。
肝細胞における不均衡
肝臓においても、高インスリン血症はFOXO1シグナルを部分的に歪めます。正常な状態では、インスリンはFOXO1を抑制してグルコース-6-ホスファターゼ(G6Pase)やPCK2の活性を低下させ、糖新生を抑えます。同時に、グルコキナーゼ(GK)を活性化して糖分解を進めます。
インスリン抵抗性と高インスリン血症が持続すると、G6PaseやPCK2は十分に抑制されて糖新生は一見低下するものの、過剰に活性化されたグルコキナーゼを介して、解糖系への炭素流入が異常に増加します。この過剰な炭素骨格はアセチルCoAの供給源となり、SREBF1を介したデノボ脂質合成を爆発的に亢進させます。
これが、肝臓における脂肪沈着と、超低密度リポタンパク質(VLDL)などのアテローム性リポタンパク質の過剰分泌へとつながります。
さらに、インスリン作用が糖代謝を抑制するのに必要な濃度(低ナノモルレベル、数分単位の迅速な作用)と、脂質合成を活性化するのに必要な濃度(高ナノモルレベル、数時間単位の持続的な作用)には、明確な用量・時間依存性の乖離が存在します。高インスリン血症という「持続的な高濃度インスリン曝露」は、まさに脂質合成経路を恒常的にドライブするのに最適な環境を提供してしまっているのです。
炎症、小胞体ストレス、そしてエクソソーム:複雑に絡み合う上流因子
高インスリン血症を起点とする悪循環の周囲には、多彩な細胞内・細胞外因子が関与して病態をさらに複雑化させています。
慢性炎症とTreg細胞の役割
脂肪組織に浸潤する免疫細胞は、TNFαやIL-6といったサイトカインを放出し、JNKやIKKβなどのキナーゼを介してIRS1の阻害的リン酸化を引き起こします。特に注目されるのは、脂肪組織に特異的に存在するPPARY発現型制御性T細胞(Treg細胞)の存在です。Treg細胞はインスリン感受性の維持に不可欠であり、チアゾリジン薬によるインスリン感受性改善効果を媒介する重要なプレーヤーであることが判明しています。また、IL-33がこれらの免疫環境を精緻に制御していることも明らかになりました。
小胞体ストレス応答(Unfolded Protein Response;UPR)
過剰な栄養流入とインスリンによるタンパク質合成の要求亢進は、小胞体(ER)に過度な負荷を与え、小胞体ストレスを引き起こします UPRの活性化は、インスリン受容体のシグナル阻害や、ミトコンドリアとの接触領域(MAM)の構造変化を誘導し、インスリン分泌能の低下とインスリン作用の障害を同時に引き起こす共通の病態基盤となります。
エクソソームによる臓器間ネットワーク
脂肪細胞は、多種多様なマイクロRNA(miRNA)を内包したエクソソームを放出し、遠隔臓器のインスリン感受性を制御しています。例えば、脂肪由来のエクソソームはFGF21の分泌や機能を抑制し、インスリン抵抗性を悪化させます。逆に、脂肪組織のマクロファージから放出されるエクソソーム中のmir155(PPARYを標的とする)やmir690(NADキナーゼを標的とする)は、全身のインスリン感受性をダイナミックに変調させています。
ミトコンドリアダイナミクスと加齢によるIgG蓄積
インスリン抵抗性組織では、ミトコンドリアの酸化的リン酸化の低下、いわゆる代謝の柔軟性の喪失(
metabolic inflexibility)が観察されます。これには、低分子量GTP結合タンパク質であるRALAの活性化に伴う、DRP1依存的なミトコンドリアの断片化が深く関与しています。また、近年の新しい仮説として、加齢に伴い脂肪組織内に免疫グロブリンG(IgG)が蓄積し、これがインスリン受容体シグナルを障害して加齢性インスリン抵抗性を引き起こすという現象が報告されており、カロリー制限によってこのIgG沈着が劇的に減少することも確認されています。
本研究の新規性と学術的限界(Limitation)
本レビュー論文の圧倒的な新規性は、インスリン抵抗性を「単一のシグナル経路の遮断」として捉える静的な還元主義を完全に否定し、高インスリン血症が下流のシグナル(FOXO1活性とmTOR/SREBF1活性など)をアンバランスにドライブする「動的かつ選択的なシグナル障害モデル」として再構築した点にあります。さらに、DAG・PKC仮説の限界をヒト遺伝学データからロジカルに指摘したことは、今後の創薬プロセスにおける標的設定のあり方を根本から揺るがす重要な指摘です。
しかし、本研究が提示するモデルにも明確な学術的限界(Limitation)が存在します。
第1に、現在のインスリン抵抗性モデルの多くは、単離培養細胞や急性的な動物モデルから得られたものであり、数十年にわたりフィードバックと代償機構が複雑に絡み合って進行するヒト2型糖尿病の慢性的な時間経過を完全には再現できていません。
第2に、2型糖尿病患者から採取した膵島組織を用いた遺伝子発現解析(トランスクリプトームなど)を行っても、全患者に共通する明確な疾患感受性遺伝子のシグネチャーを同定することは極めて困難であり、病態の背景に存在する著しい異質性(ヘテロジェニティ)が分子標的薬の開発を阻んでいます。
第3に、多くの前臨床試験で劇的な治療効果を示した候補物質が、ヒト臨床試験において有効性や安全性を担保できずに脱落するという、トランスレーショナル・ギャップがいまだに解消されていません。
明日の臨床と研究に活かす実践的アプローチ
この精緻な分子病態を理解した私たちは、明日からの臨床や研究においてどのような行動を起こすべきでしょうか。具体的な実践アプローチを提案します。
- 空腹時インスリン測定の積極的活用
HbA1cや空腹時血糖値が正常範囲内であっても、その背景で代償性の高インスリン血症が進行しているステージ(糖尿病前症)を見逃さないことが極めて重要です。定期的な空腹時インスリン濃度の測定、あるいはHOMA-IRの算出を臨床ルーティンに組み込み、シグナルの「selective insulin sensitization」が始まる前の極めて早期の段階で積極的な介入(炭水化物の質的改善や有酸素・レジスタンストレーニングの組み合わせによる代償性分泌の抑制)を行うべきです。 - 減量治療における筋肉量維持の徹底
現在、GLP1受容体作動薬やGIP/GLP1受容体作動薬による極めて強力な減量治療が主流となっています。しかし、これらの薬剤はインスリン抵抗性の根本原因である分子シグナルの障害自体を直接修復するわけではありません。さらに、急速な体重減少に伴い骨格筋量が低下すると、糖の最大のクリアランス経路を失い、長期的な代謝の柔軟性を著しく低下させる危険性があります。特に、アジア人種のように筋肉量がもともと少なく肥満度がマイルドな患者に対しては、アミノ酸や良質なタンパク質摂取の推奨、レジスタンス運動の処方を治療の初期段階から厳密に併行し、代謝的に健康な減量を達成する必要があります。 - 選択的感受性改善を標的とした創薬アプローチへの着目
従来のPPARY作動薬は極めて優れたインスリン感受性改善効果を持ちながらも、心不全、骨損失、体重増加といった副作用により臨床からほぼ淘汰されました。今後の研究・創薬においては、脂質合成や骨への悪影響を及ぼさずに、糖新生の抑制や糖取り込みシグナルだけを精密にブーストする「選択的FOXO1阻害薬」や「第二世代の選択的PPARY活性化薬」、あるいは神経系を介して代謝を改善するFGF19やFGF21の誘導体といった、次世代の治療選択肢に焦点を当てた臨床データの蓄積が望まれます。
参考文献
Domenico Accili, Zhaobing Deng, & Qingli Liu. (2025). Insulin resistance in type 2 diabetes mellitus. Nature Reviews Endocrinology, 21(7), 413-426.https://doi.org/10.1038/s41574-025-01114-y

